
Scientific data for BIOS-SCOPE will be available soon via our data partners:
.
he following are examples of the data products that have been produced recently using BIOS-SCOPE data:
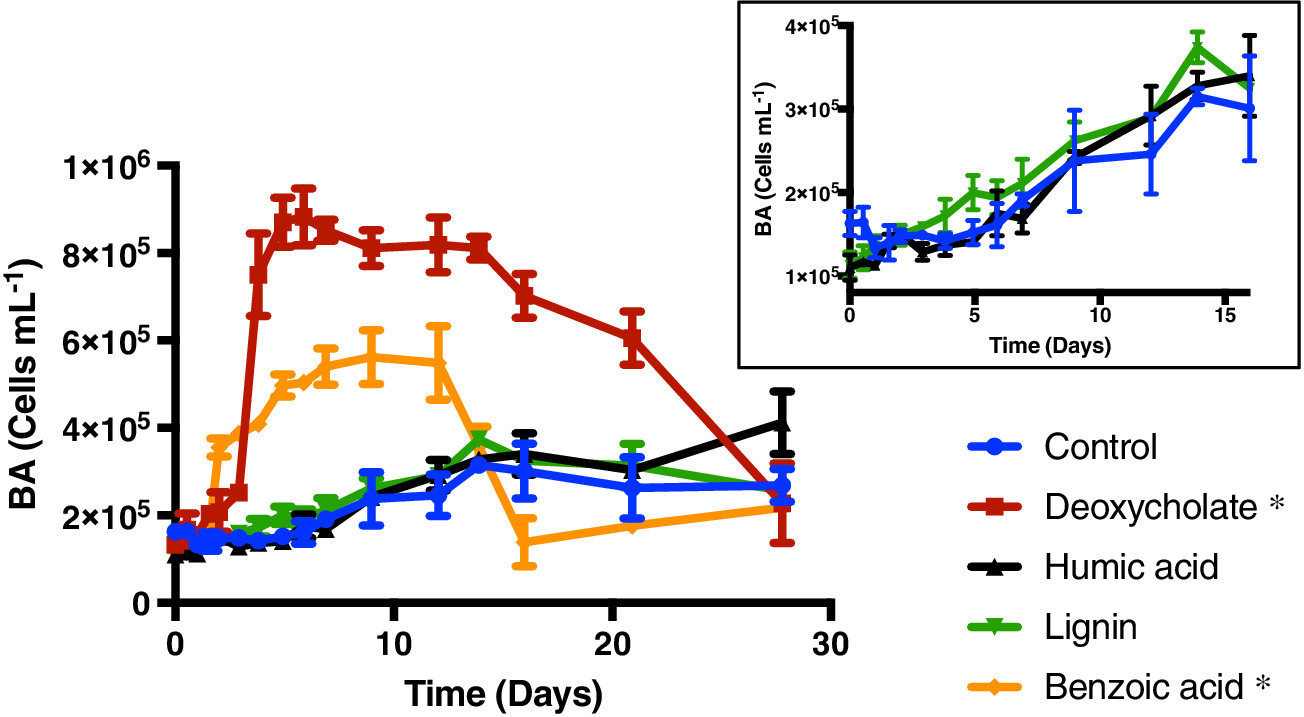
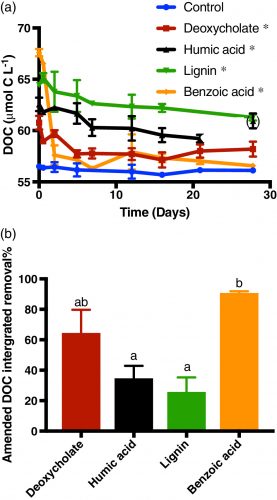

Marine sinking particles transport carbon from the surface and bury it in deep‐sea sediments, where it can be sequestered on geologic time scales. The combination of the surface ocean food web that produces these particles and the particle‐associated microbial community that degrades them creates a complex set of variables that control organic matter cycling. A recent study by Johnson et. al. (2019) published in Limnology and Oceanography used targeted metabolomics to characterize a suite of small biomolecules, or metabolites, in sinking particles and compare their metabolite composition to that of the suspended particles in the euphotic zone from which they are likely derived. The figure above shows fluxes of metabolites on the sinking particles collected in the net traps at 150 m. Only metabolites with a flux > 1 nmol m−2 d−1 in at least one sample are shown. K indicates samples from cruise KN210‐04 whereas A indicates samples from cruise AE1409. Stations are presented in order of latitude from south (left) to north (right). AMP, adenosine 5′‐monophosphate; HBA, 4‐hydroxybenzoic acid.
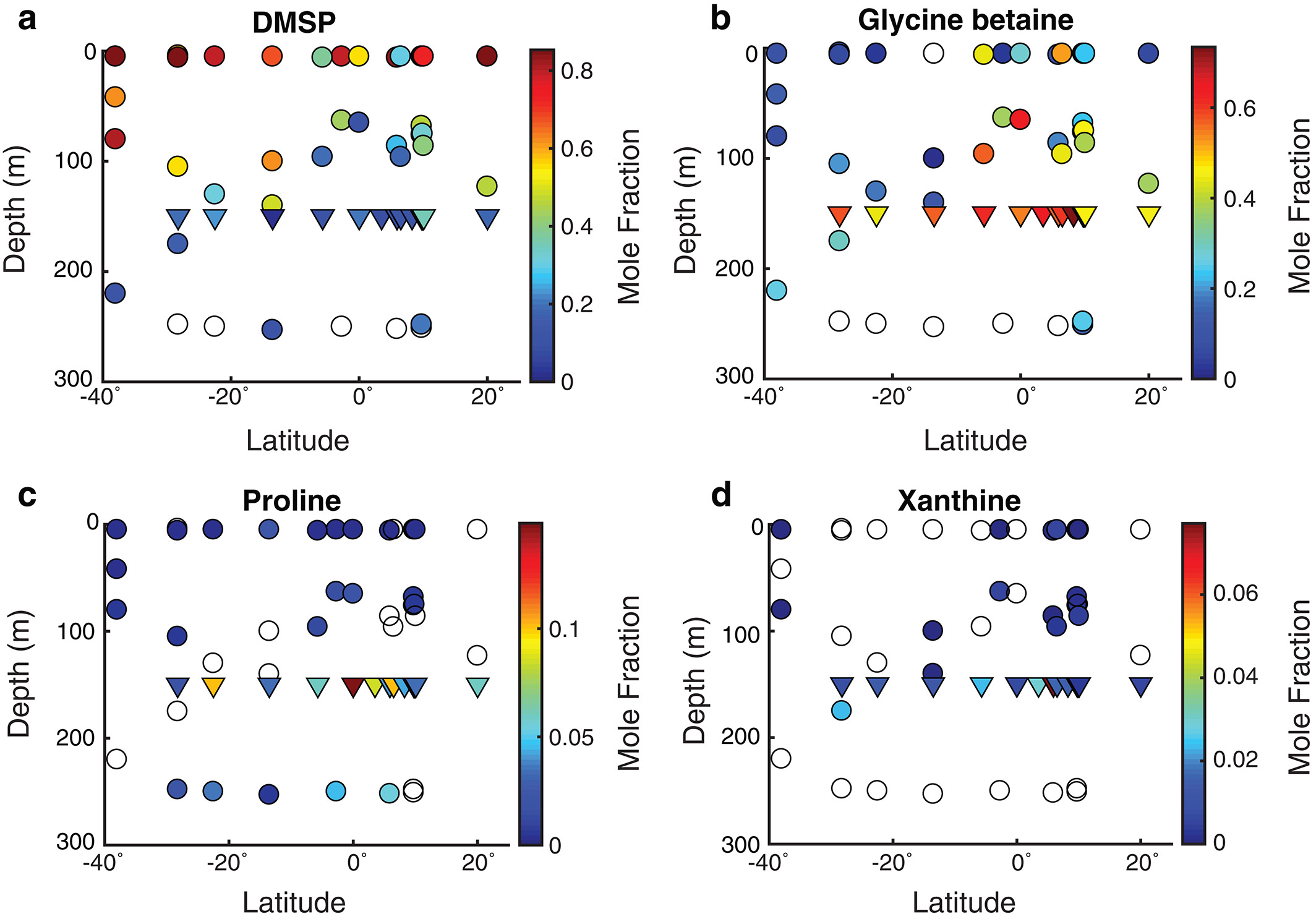
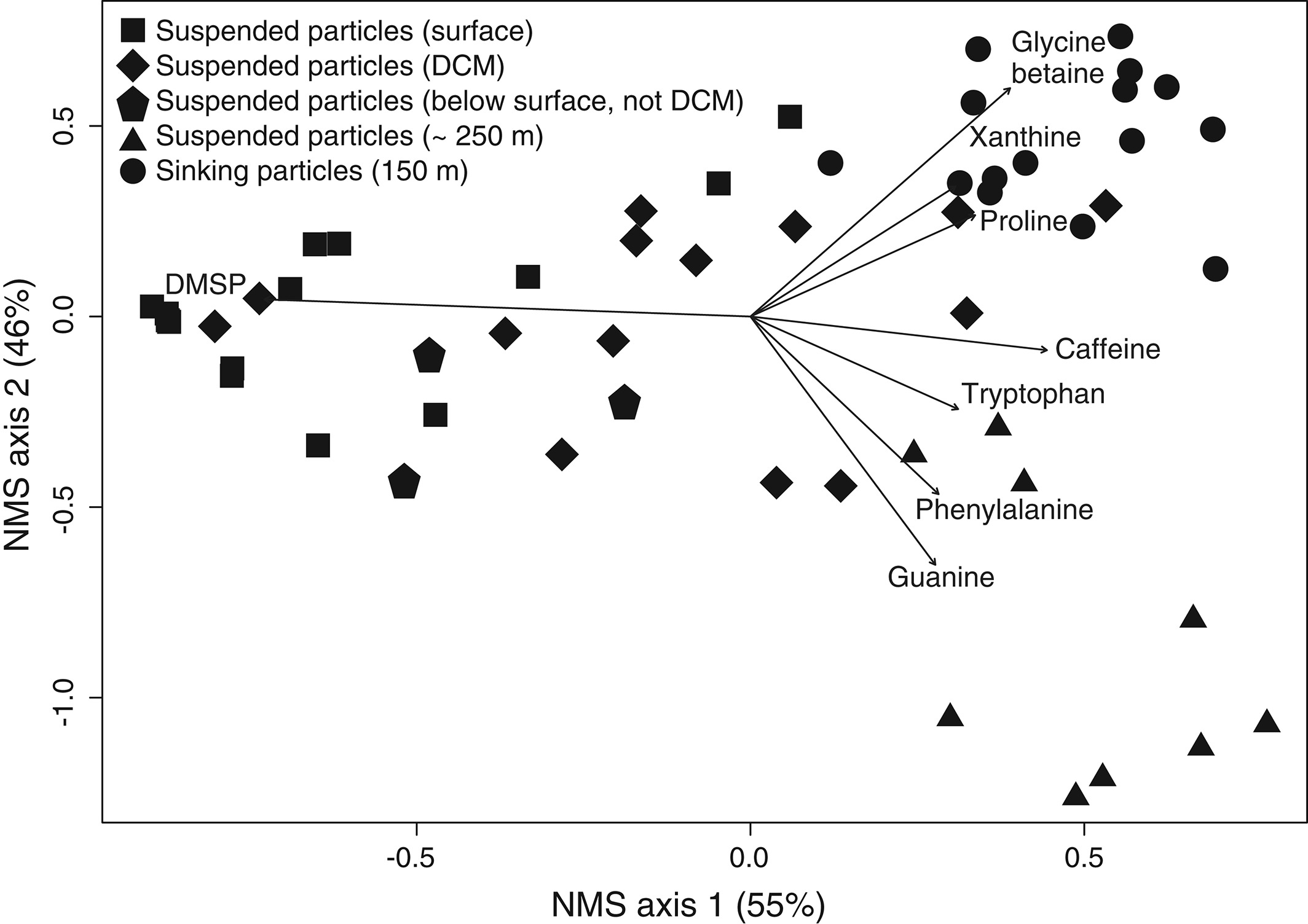
The figure above shows metabolite profiles in both the suspended (circles) and sinking particles (triangles). Open symbols indicate samples where the metabolite was not detected. Warmer colors indicate a higher mole fraction of that metabolite (metabolites normalized to total moles targeted metabolites) in the sample. Shown are (a) DMSP, (b) glycine betaine, (c) proline, and (d) xanthine. The study showed that the metabolite composition of sinking particles throughout the western South Atlantic gyre, equatorial region, and tropical North Atlantic was similar compared to the metabolite composition of suspended particles. These findings provide a new lens through which to observe microbial community activity, the processing of organic matter on sinking particles, and ultimately carbon flux to the deep ocean. While these small molecules do not comprise the bulk of organic matter transported on sinking particles, they are the conduit through which larger organic molecules are processed.
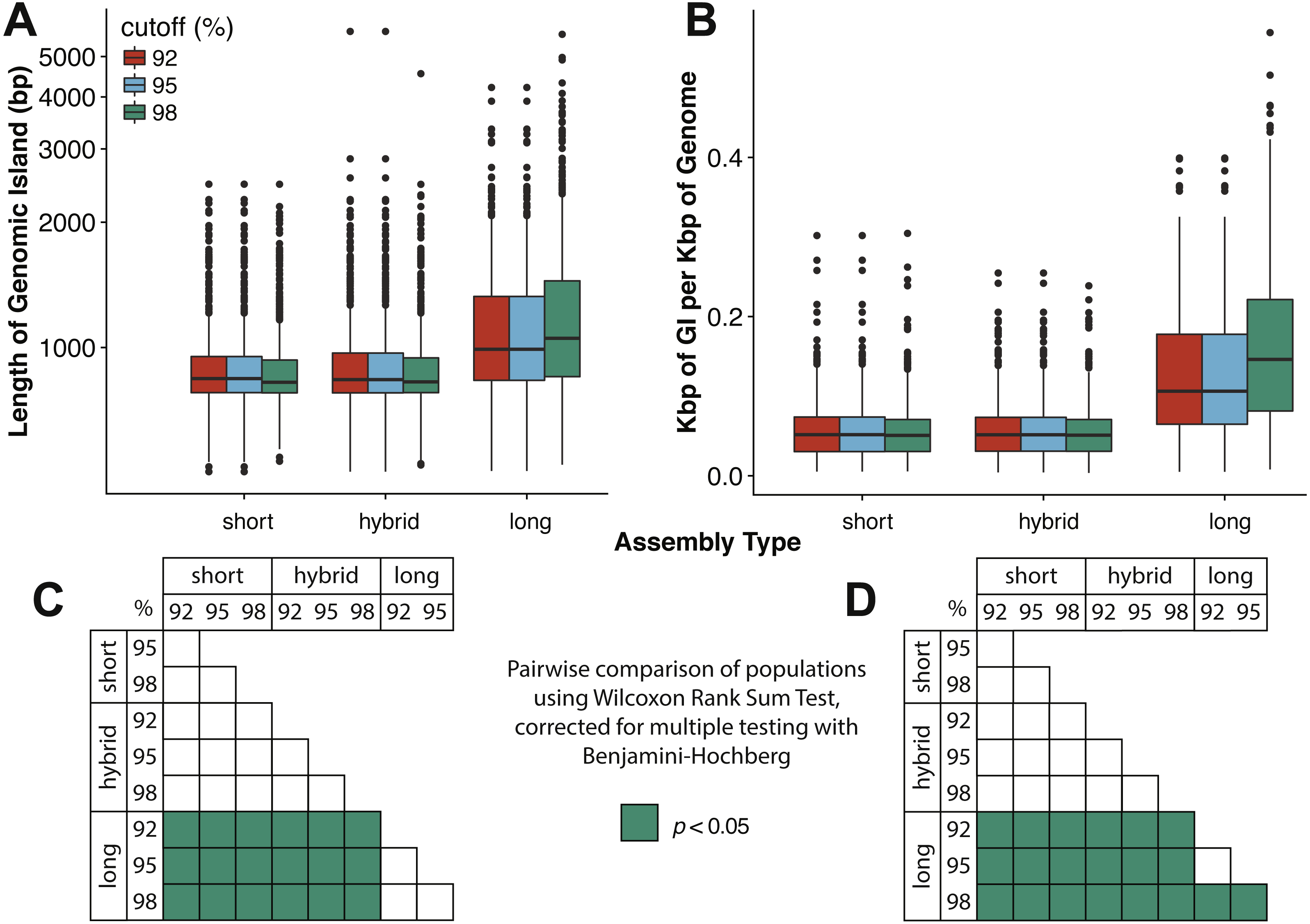
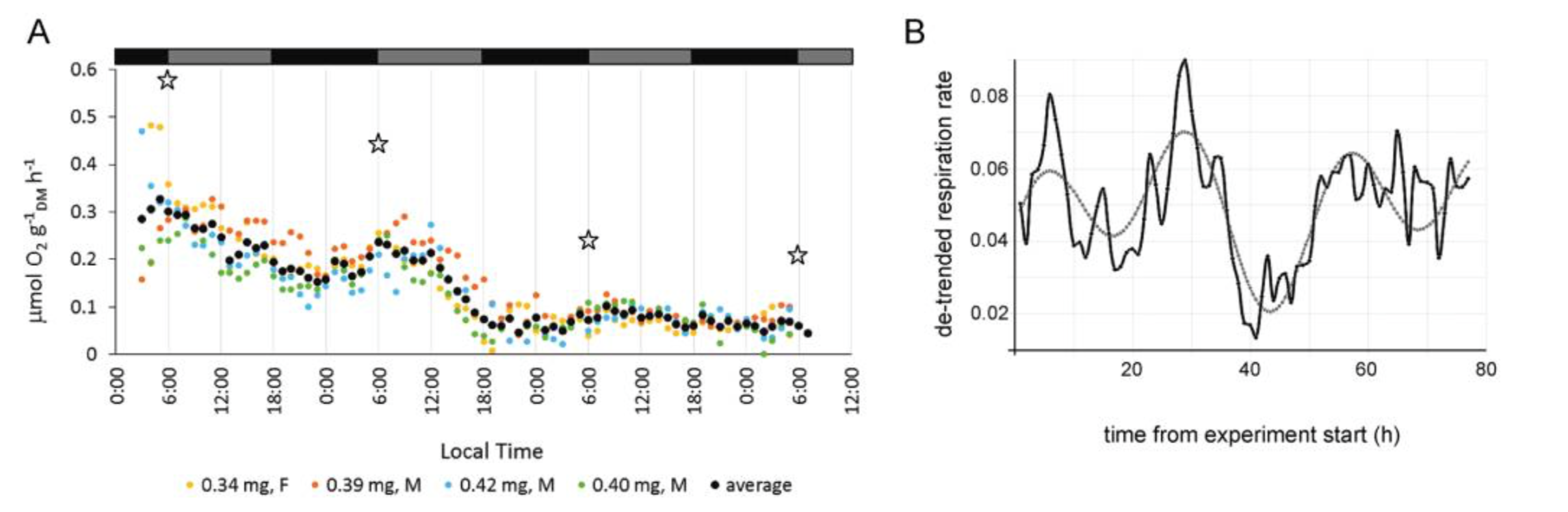
Marine viruses impact global biogeochemical cycles via their influence on host community structure and function, yet our understanding of viral ecology is constrained by limitations in host culturing and a lack of reference genomes and ‘universal’ gene markers to facilitate community surveys. Short-read viral metagenomic studies have provided clues to viral function and first estimates of global viral gene abundance and distribution, but their assemblies are confounded by populations with high levels of strain evenness and nucleotide diversity (microdiversity), limiting assembly of some of the most abundant viruses on Earth. A publication by Warwick-Dugdale et. al. (2019) in PeerJ established a low-cost, low-input, high throughput alternative sequencing and informatics workflow to improve viral metagenomic assemblies using short-read and long-read technology. The figure above shows comparative performances of short-read and long-read data for the identification of marine viral genomes (full caption in paper).
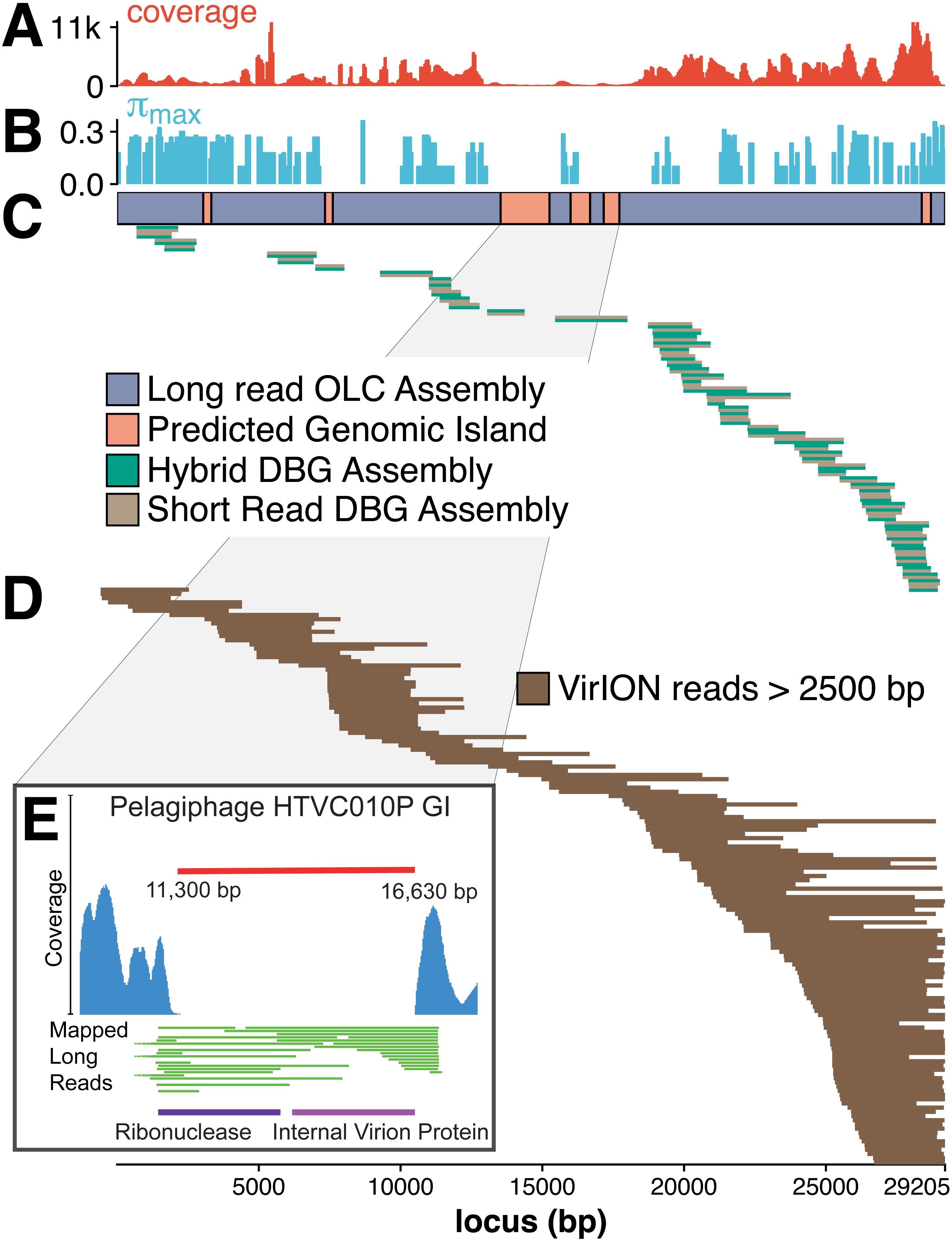
The figure above shows that long-read sequencing resolves microdiversity and assembly issues across genomic islands in ecologically important viral taxa (full caption in paper). This investigation represents the first use of long-read sequencing for viral metagenomics. Investigators showed that using long-reads to scaffold short read De Bruijn Graph assemblies improves recovery of complete viral genomes. Furthermore, overlap-layout consensus assembly of VirION reads, followed by error correction with short reads captures abundant and ubiquitous viral populations that are missed (possibly as a result of genome fragmentation) by current short-read metagenomic methods. By combining these two approaches, the proposed bioinformatics pipeline maximises the capture of viral diversity whilst minimising the impact of high error rates associated with long-read sequencing and represents a major addition to the viral metagenomics toolset. This approach represents a significant advantage in terms of cost, yield and efficiency over fosmid and single-amplified genome approaches to capturing marine viruses that are otherwise challenging to assemble.